
E&L Concepts
E&L Concepts
Concepts in Leachables Risk Management

The ELSIE consortium has developed this website to describe how extractables and leachables evaluation related to drug product (including drug-device combination products) and manufacturing systems can be approached within a risk management framework, based on the comprehensive risk management framework described in ICH Q9, and illustrated in Figure 1.
These webpages describe conceptually how each of the main elements of risk management noted in the figure – screening and materials selection, risk assessment, risk control and risk review – could be applied to overall leachables management. These are considered “living” pages and will be updated from time to time with new insights and information.
We also note and recognize several standards, industry publications, and regulatory guidelines that provide invaluable information on how to consider extractables and leachables testing and aspects of risk associated with leachables. Some of these have served as foundational documents for ways to think about leachables risk and control (e.g., PQRI recommendations for orally inhaled and nasal drug products; FDA packaging guidance; EMA plastic packaging guideline).
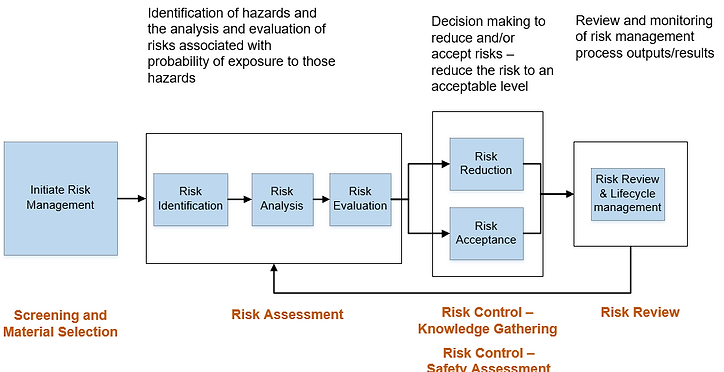
Figure 1. Risk management framework for extractables and leachables, based on ICH Q9, Quality Risk Management
Concepts in Leachables Risk Management
There are important gaps in the current global regulatory guidance on extractables and leachable. A key general gap is the lack of an articulation of leachables management that is grounded in an overall risk management framework, and which provides risk-based context within which existing standards, industry practice documents, and regulatory concepts can be prioritized and applied.
Additionally, but related to this overall gap are some specific gaps, which include:
Lack of a shared glossary of terms which can be applied in all scenarios in which there is a risk of leachables (manufacturing, small and large molecules, delivery devices, etc.)
No agreed requirement for material selection criteria, in alignment with quality by design
No broadly agreed definition of “pharmaceutical grade material”
No minimum standards or selection rules based on risk from materials
No agreed weighting of relative importance of different information sets available to define a material’s suitability with respect to leachable risk
Lack of alignment on role of supply chain quality in maintaining quality assurance through product lifecycle
No clear risk management requirement for each dose form type (requirement most clear for high risk dose forms only)
No coherent and consistently described process for risk management of leachables in any guidance which is applicable to all sources of leachables (including manufacturing, packaging, and administration) covering all modalities
No agreed definitions for risk factors (Severity, Probability) to allow risk analysis and scoring
No guidance on an appropriate risk evaluation matrix for transformation of risk analysis into risk control decisions
Lack of clarity and consistency in regulatory expectations for risk control at each stage of drug development and through the lifecycle
No agreed study design requirements for each study type (extractables and leachables)
No holistic, unified guidelines that consistently apply the threshold concepts of reporting, identifying and qualifying impurities.
No agreed safety risk assessment limits for all toxicological end-points
Lack of defined process for study of lifecycle changes and risk review
No definition of Major and Minor changes which might affect leachables
No alignment to requirement for prior approval or post notification change
The content on these webpages will refer to some of these gaps within the context of the proposed comprehensive risk management approach, and its specific elements.
If you have feedback to these pages, please contact us.