
Screening and Materials Selection
Screening and Materials Selection

Introduction
Materials and components used in the manufacture, storage and use of pharmaceuticals (small molecule and biologics) and devices in drug delivery systems should be evaluated for extractables and leachables (E&L) where they are in direct contact with the active pharmaceutical drug substance (DS), drug product (DP), process fluids or indeed in direct contact with the patient (ICH Q7, 21 CFR 211.65(a)). Thus, it is key and reflective of regulations that, in order to reduce risks associated with some leachables, pharmaceutical companies, in collaboration with material and component suppliers develop and use an appropriate knowledge base for screening and material selection for products in development and during lifecycle decision making.
This paper describes an approach for early and subsequent assessments of materials and/or components under consideration for use in pharmaceutical active drug substances (DS) and drug product (DP) manufacturing processes, container closure systems, and drug delivery devices. This approach may be considered as a “hazard appraisal process” or HAP, which may be used within a broader ICH Q9-type risk management process. The HAP can assist in understanding key aspects of a screening and/or materials selection process such as what data is available; how to gain access to it; assess its weighting to the scenario in hand; how to build an informed hazard assessment or profile in a consistent manner to then communicate with stakeholders.
An example decision process flow is provided to capture the elements of a HAP, with the inclusion of six case studies for materials and component screening to exemplify its potential use with a diversity of scenarios, to reasonably reflect current pharmaceutical development and lifecycle practices.
Gaps and Challenges
Some current gaps and challenges that face pharmaceutical manufacturers specifically with respect to executing an effective material and component hazard assessment include but are not limited to:
Absence of a common regulatory framework for assessment, control and lifecycle management of E&L, which includes consideration of screening and materials selection.
Absence of globally aligned safety thresholds.
Limited access to supplier data, the data being non-contemporaneous in nature or non-standardised in structure.
The wide diversity of conditions (be they extractables or leachables studies) under which E&L data is collected can make extrapolation to specific scenarios more challenging.
The overall large diversity of potential E&L that could be present for a given material/ component and conditions of use, and the absence of a common accessible knowledge base.
Articulation of a rationalized process, such as a HAP, for screening and materials selection, within a larger risk management process for leachables, can help address some of these opportunities, responsibilities and challenges.
Within a typical risk management framework, such as that noted in ICH Q9, the screening and/or materials selection activity may be envisioned to occur prior to risk assessment (highlighted at the beginning of Figure 1) and may be applicable in a new product development or lifecycle management scenario.
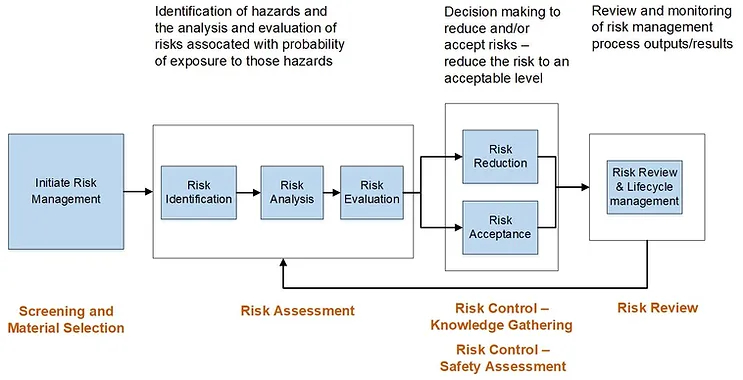
Figure 1: Risk management framework for leachables, based on ICH Q9 risk assessment process.
A HAP for screening and materials selection provides a framework to collate and classify existing knowledge providing a consistent and considered output which could then feed into a formal technical product-based risk assessment process. This approach is consistent with general risk management principles including the identification of hazards, evaluation of the risks followed by mitigation and control. A HAP facilitates a structured review of appropriate materials and components, and informed decision-making as to general fitness of materials and components for their intended purpose and stage of development. Note that a HAP is not the same as risk assessment and risk control, but rather entails evaluation of supplier information and prior knowledge to inform these later steps in the risk management process..
Scope
Materials and components in scope include those used in manufacturing process systems, those in direct contact with DS or DP during bulk storage and as part of packaging, container closure systems (CCS) or a drug delivery device, as well as secondary packaging. As per USP, “materials” are materials of construction, and “components” are packaging components (USP <659>, Packaging and Storage Requirements).
The topic is relevant throughout the development and commercial stages of pharmaceutical products (including biologics) and drug delivery devices, thus there may be partial convergence with aspects of lifecycle management and change control relating to E&L. The philosophies described here could be considered for use in advance of the anticipated ICH Q3E guidance on E&L which may apply to commercial products as well as those in development.
Materials and components not in scope would generally include those that do not come into direct contact with the DS, DP or process fluids. Medical devices such as pacemakers, stents, etc. are out of scope. General considerations for design of experiments or generation of actual data are also out of scope as these are covered in risk control- knowledge gathering.
Figure 2 describes an E&L hazard appraisal framework concept and the key aspects that would be explored by a company as it gathers existing knowledge and undertakes an initial hazard assessment. This knowledge gathering and science and risk-based control strategy can evolve alongside the development lifecycle of a medicinal product. Key considerations would include the full utilization of existing regulatory guidelines and pharmacopoeial standards and leveraging other information including historical data sets, published studies, and data modelling.
Figure 2: Conceptual representation of an E&L Hazard Appraisal framework.
Elements of Hazard Appraisal Process
Fundamentally, the E&L HAP framework presented utilizes existing approaches and established principles. These include key considerations such as route of administration, prior safe use, likelihood of CCS interaction, the manufacturing process, and the magnitude and duration of exposure to the patient be it via the DS or DP. The main elements of the HAP process flow are shown in Figure 3. This includes some key questions that should be traversed for any E&L assessment, for example around potential contact with the DS or DP and whether the material or component will be part of the container closure system or the manufacturing process.
Figures 5, 6, and 7 (Appendix 1) depict the hazard “grids” relevant for manufacturing, route of administration and CCS interactions. These grids essentially provide a scaffold or prompts around which a hazard level can be assigned, they have a basis in either regulatory guidance or general Pharma industry practice.
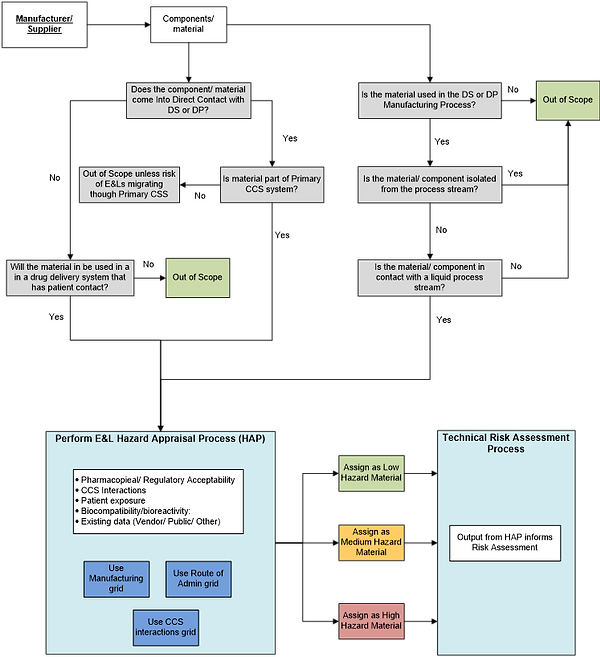
Figure 3: Summary process flow for screening materials and components with respect to extractables and leachables management.
Thus, Figure 3 above and the hazard grids (Figures 5-7 in Appendix 1) provide a suggested framework through which pharma could review available data, and make consistent, informed decisions on the initial hazard category that new components or materials can be allocated from an E&L perspective and, where multiple component or material options are available, allow an initial relative ranking. For precedented / existing / previously used components or materials, the framework remains applicable with the expectation that the hazard appraisal would be mitigated by a larger applicable dataset (e.g., “use data”).
As mentioned above the process flow in Figure 3 provides an example set of initial questions (grey boxes) to identify only those components or materials that require an assessment HAP.
Existing information on the materials/ components and required compliance/ safety aspects can be gathered from a diversity of sources, examples of which are summarized in Figure 4. Appendix 2 provides a more extensive list of references. These represent sources of data that can feed into the relevant elements of the HAP.
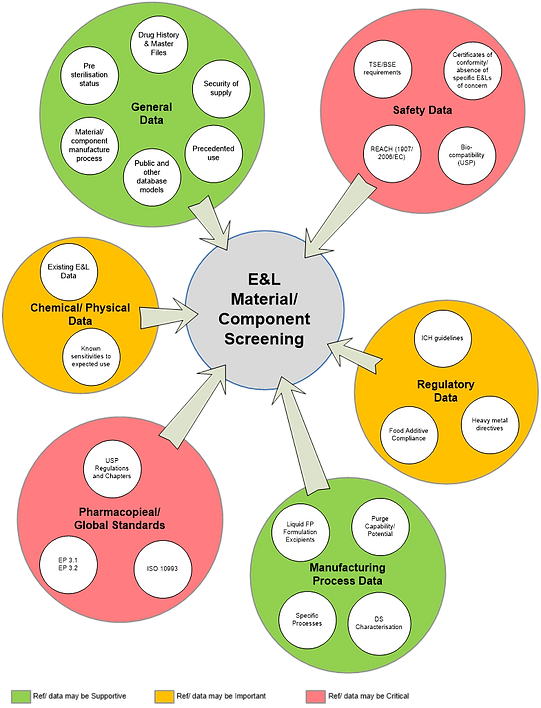
Figure 4: Examples of E&L information sources for material and component screening.
Regulatory (Compendial) Requirements
For new materials or materials subject to change control, there are various regulatory regulations, pharmacopoeial and other standards in place. For example, USP, for plastic materials and elastomers intended for use in pharmaceutical environments (see Appendix 2). The draft USP <665> in conjunction with <661.1> and <661.2> provides an appropriate “suitability for use” framework to evaluate plastic materials under consideration for use, solely targeting processes involving liquid streams. Other comparable standards such as Ph. Eur. chapters 3.1 and 3.2, and ISO are also in place. Refer to Appendix 2 for a comprehensive listing.
Evidence of this compliance (including characterization) should be provided by the material or component manufacturer. Where materials and components subsequently change, to maintain control and ensure the risk profile does not increase, the manufacturer and/or suppliers can work to ensure that the appropriate change control process has been completed with evidence of continuing compliance against standards being provided to the pharmaceutical company. Overall compliance allows a low hazard assignment to the materials/ components. For commercial products data should be generated to confirm regulatory compliance. The default would be testing against the current pharmacopeial standards (e.g., USP, Ph.Eur.).
Alternative sources of data may be utilized to confirm regulatory acceptance with the appropriate scientific rationale and hazard evaluation. This could include but is not limited to food contact compliance, manufacture’s data, comparator data, etc. During the development cycle a formal technical risk assessment should be undertaken and the appropriate data generated.
Food Contact Requirements
Where pharmacopeial data unavailable or limited in early development, as a minimum materials and components should meet the requirements of current regulations such as (EC) No 1935/2004, (EU) No 10/2011 and amendments and 21CFR Parts 172–179. Where materials and components subsequently change, to maintain control and ensure the hazard profile does not increase, the manufacturer and suppliers should ensure that the appropriate change control process has been completed with evidence of continuing compliance against the standards being provided to the pharmaceutical company.
Certificates of Conformity and Absence of Specific E&Ls of Concern
Available evidence should be gathered to confirm absence or assurance below current permissible tolerable daily intake (e.g., 4 mcg/kg/day or lower) for special case E&L such as BPA, PAHs, PNA’s MCP, N-nitrosamines, nitrites, etc. The expectation is that the supplier or manufacturer will provide the appropriate conformity statements to Pharma.
Existing CRO/ other E&L Data
Any other information, in additional to evidence of compliance, relevant to E&L should be requested from vendor/supplier and CROs and/or gathered from internal company sources. This can be a broad range of information including vendor DMF (drug master file) or DHF (design history file), consortium and public E&L data. It may indeed also include modelling information.
Examples of Hazard Grids
Container Closure System Interaction Hazard Grid (Figure 5, in Appendix 1)
The FDA’s Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics Chemistry, Manufacturing, and Controls Documentation, provides a framework against which an assessment of the CCS interaction hazard can be determined. This is a key area for consideration for a HAP given the relative contact duration and in terms of DP lifecycle, the relative proximity to the patient.
Exposure Duration Hazard Grid (Figure 6, in Appendix 1)
Consideration of the anticipated exposure profile to the patient should be undertaken with specific consideration of the dosing regimen. Again, this is a key area of consideration for a HAP particularly for chronic dosing of drug products. Single acute dosing presents a relative low hazard while the hazard will be increased where chronic dosing is required. Precedented use of these components in comparative scenarios may be used to form part of the assessment following the principles within the draft USP <665>. The principles of ICH M7 (small molecule) and ISO-TS 21726:2019 (devices) can be adopted for and provide a relevant framework and tables for the HAP assessment and the hazard category for exposure.
Manufacturing Interaction Hazard Grid (Figure 7, in Appendix 1)
A review of the expected DS and/or DP manufacturing process should be undertaken to understand the impact of direct contact materials and components to the DS or DP with respect to E&L, as these process equipment related leachables may affect CQAs of the DP or present a safety hazard to the patient by their presence in the DP. A diverse range of conditions may impact the manufacturing E&L hazard (e.g., sterilization or high temperatures, solvent type, storage, duration, etc). Comparable data will greatly facilitate understating of the manufacturing hazard.
Biocompatibility Testing
While biocompatibility testing is strictly not encompassed within the assessments of E&Ls, there is significant overlap in terms of patient safety considerations. Some materials have the potential to come into direct or indirect contact with the drug product and the patient and thus should be also assessed for their potential to trigger an adverse biological response. These are typically materials used in a CCS component that contacts the drug product during storage/ use and then could come into contact (i.e., skin, mucosal, etc.) with the patient during administration of DS or DP. Biocompatibility can be deemed a material property, indicating a baseline level of acceptability for its intended use with respect to toxic, injurious, or immunological responses in living tissues. Thus, when evaluating materials or components during the early screening and selection process, consideration should be given to type of body contact, duration of patient contact, clinical/commercial use, and total surface area of the material/component. There are various standards and guidelines in the public domain that address biocompatibility testing including the globally recognized ISO 10993 standard for biological evaluation of medical devices (see Appendix 2).
Case Studies
There is a large diversity of E&L scenarios that may present to pharma during development. The six case studies presented here attempt to exemplify that diversity, with a spread of components/material, hazard category and across the development life cycle. For each, the available information is assessed following the thought processes outlined in the decision trees and relevant elements of the HAP.
Overall, the case studies arguably demonstrate the use of HAP as an approach for E&L subject matter experts to structure and communicate a hazard assessment of materials or components with development teams and to provide some guidance on next steps. The output is an example to justify using or not using materials/ component, to proceed to use if additional information/supportive evidence is generated as part of a product specific technical risk evaluation or indeed to rank order several materials/ components. In actual application, the process and output will vary be case by case.
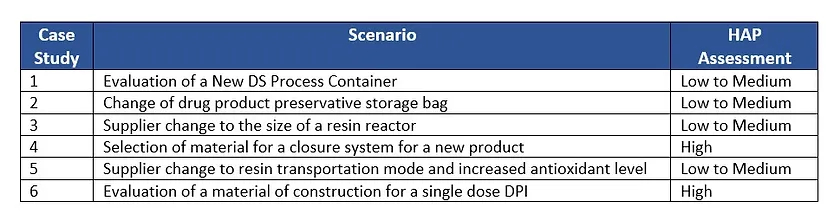
Below is a list of the six case studies provided and their HAP assessment.
The studies are largely based on real cases but will include some hypothetical data to better convey the scenario.
Case Study 1: Evaluation of a New DS Process Container
Background
As part of the manufacturing process of a small molecule, a process stream of the final, non-isolated DS requires transfer from one location to another for final isolation. A couple of options are under consideration for the holding and transferring of the solution, one of which is the use of a semi rigid single use process container, which is manufactured from a tri-layer film containing LDPE as contact layer, polyester outer layer and an adhesive tie layer in-between.
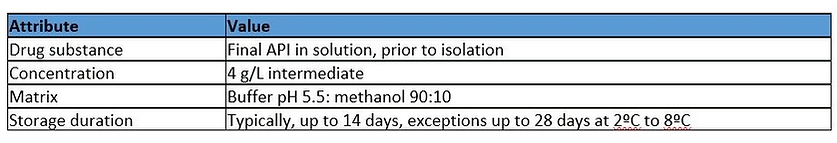
Relevant drug product Quality Target Product Profile (QTPP) Attribute
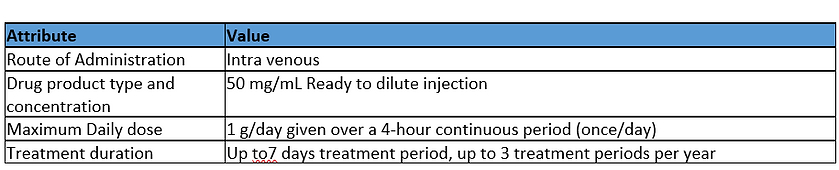
Assessment
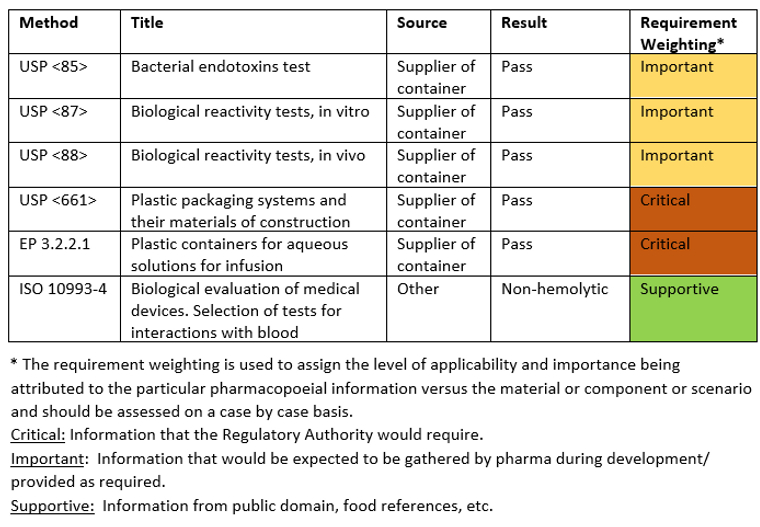
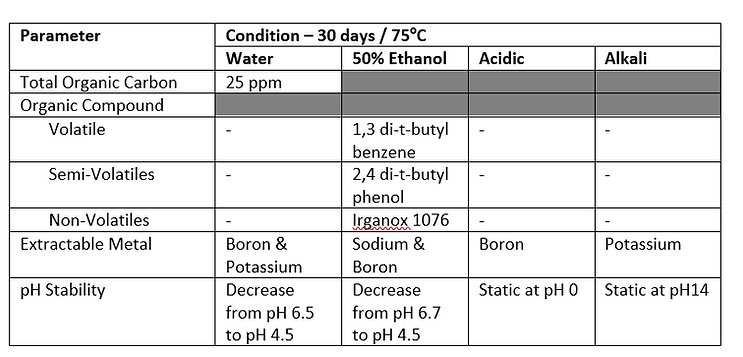
Existing Extractables and Leachables Data review
Note: This data is a compilation of supplier, internal and other information, including E&L levels, SCT values, etc. Thus, the format/ content type will vary for each scenario.
Precedented Use and Existing Leachables Data
While there is no specific extractables or leachables information available internally on the use of this specific type of container or material, there is an extensive body of data (both internally and externally) available on polyethylene as contact material which demonstrates precedence and gives confidence in the potential use of this material in this application and would be expected to present a low leachable hazard.
Hazard Appraisal Process
Pharmacopoeial Requirements: Low Hazard
The above weightings have been applied to demonstrate the criticality of this data based on the intended use of the containers. Supplier information has demonstrated that the plastic container material has met the main pharmacopoeial requirements (USP <661> and EP 3.2.2.1), the availability of this critical information facilitates a low hazard assignment. Data is also available for other requirements which has a lower weighting. Bacterial endotoxins, biological activity tests and blood interactions that have been given a medium/low weighting due to the additional processing that the contained drug substance solution will undergo during isolation and drug product manufacture. It is recognized that the contained solution is the finished drug substance and could be impacted.
Container Closure System: High Hazard
Based on the drug substance in liquid form being in contact with the plastic container, forming a high likelihood of interaction with the packaging, and the highest degree of concern for the route of administration, the CCS HAP supports a high hazard assignment. It is noted that 1,3 di-tert-butyl benzene, 2,4 di-t-butyl phenol and Irganox 1076 have been identified as extractables from the containers. These are standard additives, routinely found as leachables in these types of materials. With the exception of 1,3 di-tert-butyl benzene (degradant), a significant body of toxicological safety data exists. While these E&L impurities are considered not to be of concern, the lack of data on the degradant indicates a potential hazard that would need to be considered further.
Exposure Duration: Medium Hazard
The dosing regimen expected for the drug product is short duration on a non-regular basis over the patient’s lifetime. Based on this information the exposure duration is given a Medium hazard assignment.
Manufacture: Low to Medium Hazard
Based on the composition of the drug substance matrix (including low percentage methanol), the duration of contact (up to 28 days) and the proximity of the container in relation to the finished drug product, there is potential for the identified compounds to be extracted. However, this is mitigated by the storage temperature (2ºC to 8ºC) for the container. Based on this data and limited information on precedented use in similar environments, the manufacture hazard assignment is Low to Medium.
Conclusion
The overall HAP assignment for the container is considered Low to Medium Hazard. It is recognized that the CCS hazard is deemed high, with due consideration and understanding of the body of data available on the potential extractables and leachables. The impact of 1,3 di-tert-butyl-benzene, 2,4-di-tert-butyl-phenol and Irganox 1076 on the end drug product, are considered low and should be evaluated during a specific technical assessment as potential leachables in the final drug product.