
Risk Review-Lifecycle
Leachables Lifecycle Management Within a Risk Management Process

Introduction
We describe here some key considerations and concepts, as well as current gaps in the regulatory framework related to leachables lifecycle management within an ICH Q9-based risk management process. In particular, we propose that the key issue in leachables lifecycle management is post-approval changes. We will thus focus on this topic, discussing how leachables lifecycle management can be incorporated into an overall risk management process for drug products and manufacturing systems, and how this risk-management framework can facilitate development of rationalized change management processes.
Key concepts and gaps noted are:
Within the context of the ICH Q9 risk management framework, lifecycle management is encompassed by “risk review” (see main Introduction). Lifecycle management will thus include consideration of earlier risk management activities, such as risk assessment and risk control.
Many lifecycle-related challenges that industry experiences with respect to leachables management occur during post-approval, specifically post-approval changes to packaging, container closure systems, manufacturing components.
There are some regulatory documents that describe general approaches to change management, e.g., ICH Q12, FDA guidance, European Commission legislation, and industry practice documents addressing leachables-related change management, e.g., BioPhorum/BPSA industry proposal.[1] However, there is currently no regulatory guidance that addresses leachables-related change management for drug products or manufacturing systems, despite the importance, impact and occasional complexity of lifecycle changes with respect to leachables. For example, there is a
– Lack of a defined process for the study of lifecycle changes and risk review
– No definition or framework to define the level of risk which might affect leachables
– No alignment to requirements for prior approval or post notification change
Regulatory Considerations – A Risk-based Approach to Classification of Changes
Various regulatory agencies as well as the International Committee on Harmonization (ICH) have encouraged industry to apply a Quality by Design (QbD) approach to pharmaceutical product development, e.g., ICH Q8, ICH Q9, the FDA process analytical technology guidance, and cGMPs for the 21st Century.[2], [3], [4] These documents provide industry with general concepts on incorporating risk assessment tools and processes into product development and lifecycle management activities.
Regarding change management specifically – the key area impacting lifecycle management relevant to leachables — ICH Q12 provides some general guidance on change management and suggests a characterization of change together with a clear understanding of how the change might affect established conditions (ECs).[5] ECs in the context of leachables might mean existing risk assessment and risk control activity including any submissions and commitments made to regulatory authority. Leachable risk and its associated activity might therefore be part of a larger project affected by the change and consideration to that requirement must be made perhaps by inclusion of leachable risk assessment and controls into a Post-Approval Change Management Protocol (PACMP). ICH Q12 breaks changes into two broad categories which are in alignment with regulatory authorities’ classifications. These are:
Prior Approval (high risk)
Notification (moderate to low risk)
Additionally, regulatory authorities in Europe and the United States have published guidance on the classification of changes to approved marketing authorization applications (MAA), and new drug applications and abbreviated new drug applications (NDA, ANDA).[6], [7], [8] Both have a common basis in which the change is classified based on its expected risk to either product safety or quality/efficacy. In Europe, that classification is a sub-division marked as Type IA, Type IB or Type II, in this classification:
Type IA is a “minor variation….that has only a minimal impact, or no impact at all, on the quality, safety or efficacy of the medicinal product concerned”
Type II is a “major variation…which is not an extension, and which may have a significant impact on the quality, safety or efficacy of the medicinal product concerned”
Type IB is a “variation which is neither a minor variation of type IA nor a major variation of type II nor an extension”
For the US, FDA guidance similarly defines Major, Moderate and Minor Changes and aligns these with the need for notification via prior approval, changes being affected supplement, or annual report.
Both sets of documents provide high-level guidance associated with the regulatory process for managing changes to registered drug products and include examples of the types of changes that fall into each of these classifications to facilitate application. Given the diversity of pharmaceutical drug products this guidance is intended to cover, and the sheer number of changes that could occur to these products, it is inevitable that there is a degree of subjectivity and inconsistency in how regulators and industry interpret the regulatory guidance for extractables and leachables with respect to manufacture, packaging, and delivery of drug products. Furthermore, there are changes that are not required to be reported to regulators but should still be assessed to establish the impact on the extractable profile of a material or leachable profile of a drug product.
Developing some specific guidance to assess the impact a change has on an extractables or leachables profile in addition to regulatory guidance that already exists, would provide industry with greater clarity on how risk management principles should be applied to ensure changes to registered drug products do not adversely impact patient safety.
A Risk Review Process Relevant to Leachables
It is essential that a risk management culture and framework is developed by industry experts in collaboration with regulatory bodies and standard setting organizations to ensure manufacturers of pharmaceutical drug products can readily categorize changes based on the level of risk in accordance with a standardized process, and be able to refer to guidance on how to address each. This will help ensure that post-approval changes are assessed in a consistent manner and promote a risk-based culture for lifecycle management where the level of scientific rigor to address the change is commensurate with the risk.
To contribute to this discussion, we describe here, considerations for a risk-based framework for lifecycle management that is aligned with the risk management concepts discussed in ICH Q9. Considering lifecycle management with respect to leachables within a risk management framework can assist in developing a risk-based rationalized process for change management. This framework is illustrated in Figure 1.
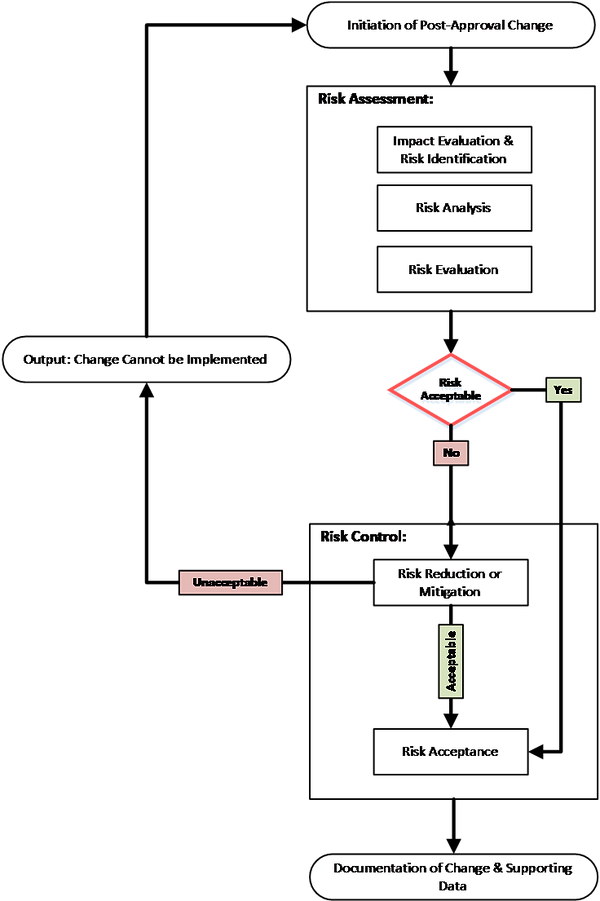
Figure 1. Risk review/lifecycle management based on ICH Q9 risk management concepts.
Initiation of Change
Background
Materials supplied to companies that manufacture food packaging systems, pharmaceutical container closure systems and delivery systems are governed by requirements that protect the end user. In the case of a pharmaceutical drug product, the end user is a vulnerable patient. Global regulatory agencies have strict requirements to which manufacturers of packaging systems and drug delivery devices, as well as final drug product manufacturers, should comply. In the United States, the Food and Drug Administration (FDA) often partners with the United States Pharmacopeia (USP) and other organizations to develop requirements that assure products meet stringent safety and quality standards. Other regions, such as Europe, Canada and Japan, have the same responsibility of protecting the end users by issuing similar standards and guidance for industry. There is evidence that regulators recognize the concepts and value of risk management practices for “new” packaging systems, and it is thus reasonable to assume that this philosophy can be extended to lifecycle management.[9], [10]
Lifecycle management of pharmaceutical container closure systems (CCS), delivery systems or single-use systems (SUS) relies on individual parties within the supply chain having comprehensive product quality systems (PQS) and supplier agreements that detail specific customer requirements since changes to a commercialized product’s manufacturing process, container closure or delivery system are inevitable. Such changes to a CCS or single-use system (SUS) can originate from within the supply chain or pharmaceutical drug product manufacturer. Examples of such changes could involve changing of the formulation grade to improve performance, making grades redundant that are no longer viable and cost-effective to manufacture, changing their manufacturing or procurement strategy to reduce costs and/or increase supply chain security. Supply agreements that address change classification as well as clear roles and responsibilities, can facilitate a rapid qualification of changes with minimal disruption to the supply of medicine to patients.
One of the many obligations that product manufacturers and their supply chain partners have for a change involving a material used to manufacture, package or deliver a pharmaceutical drug product is to assess the impact of that change to patient safety. Materials are a potential source of chemical compounds that, over time, leach into the pharmaceutical drug product. As such, changes that alter the chemical composition could promote leaching of compounds from the material into a drug product that could potentially affect the safety profile of the drug product. Some of these changes throughout a product’s lifecycle will have negligible impact on patient risk, whereas others may be more significant and need to be assessed more thoroughly by either gathering more information and/or conducting experimental studies. This process typically relies on input from many scientific disciplines, including scientists within the supply chain, toxicologists, analytical chemists, quality assurance, engineers etc. As such, the importance of the change notification process cannot be underestimated as it is critical for the efficient flow of relevant knowledge within the supply chain necessary to understand and assess the impact on the quality and safety of the finished drug product.[11]
Examples of Changes
Throughout the product lifecycle, changes to the drug product packaging, delivery systems, and manufacturing systems may require an impact evaluation with respect to extractables or leachables, and subsequent risk assessment revision. These changes could originate externally from the supplier of the part or internally from the drug manufacturer. In either case, drug manufacturers change management processes should include an understanding of the changes that impact leachables and what actions were taken to implement the change. These processes could incorporate leachables considerations established during development and supported by product knowledge, including a Quality Target Product Profile (QTPP) where available. Table 1 provides some examples of changes to single use systems and components that could be relevant to leachables management.
TABLE 1. Examples of changes relevant to extractables and leachables. (Note: this is not an exhaustive list)
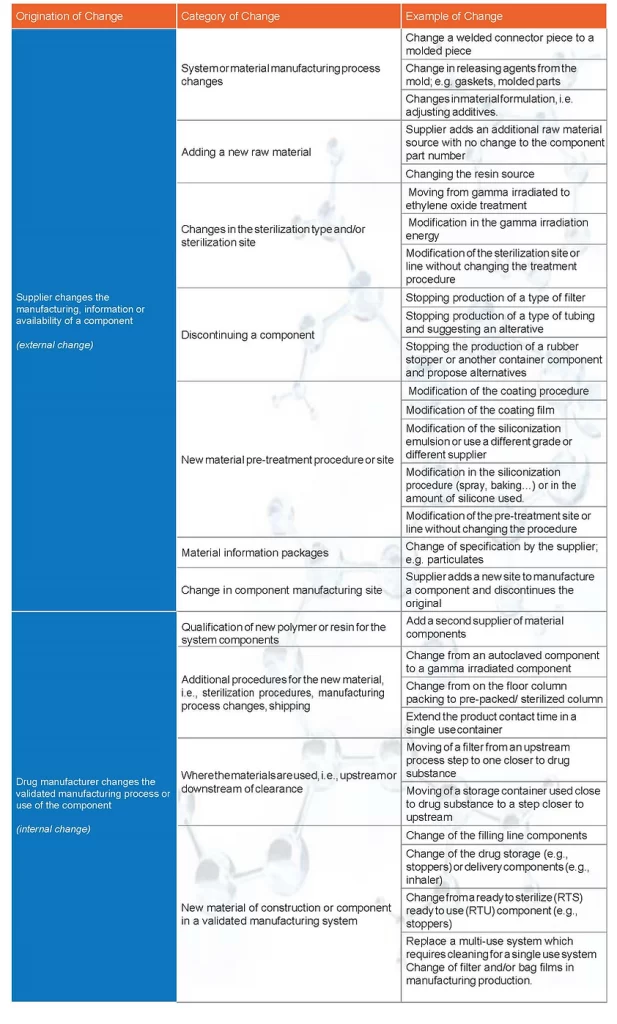
The required work to implement the change should be clearly documented in the drug manufacturer’s change management processes. Each change should be assessed independently to (i) allow the drug manufacturer to ensure the final product is comparable to historical batches and (ii) communicate the change and implementation path upon request.
Once the change is initiated and understood by the drug manufacturer, the impact of the change can be determined by following the elements of the risk assessment process, i.e., risk identification, risk analysis and risk evaluation followed by risk control. The change could require risk control activities such as re-validation, repeating of the extractable and/or leachable studies, as well as updating process validation technical packages, supporting stability studies and regulatory filings.
The following sections describe risk assessment and risk control activities within a lifecycle management framework for extractables/leachables. These concepts are described in more detail in risk assessment and risk control.
Risk Assessment
As noted in the risk assessment pages, risk assessment activities include risk identification, risk analysis and risk evaluation. These activities facilitate understanding of relative risk to the product and thus inform conclusions and decisions regarding the relative impact of a change.
Once the risks that can potentially impact leachables are identified, they can be analyzed and evaluated based on the severity and probability factors as noted in risk assessment. During the risk assessment process, knowledge that has been acquired from a variety of sources, including the supply chain, internal archives, materials scientists, project teams, manufacturing sites, etc., should be leveraged to inform the severity and probability. As noted in materials selection pages, in addition to knowledge that exists within a pharmaceutical drug product manufacturer’s organization, a large amount of knowledge is generated on products throughout the material supply chain to assure customers that a manufactured product meets the required quality and safety requirements for its intended purpose.
Manufacturers of pharmaceutical packaging and delivery systems rely on suppliers that are often global organizations having a good awareness of the regulatory expectations for materials intended for use within the food, pharmaceutical or medical device industries. They often develop their product with these regulations in mind and generate data that can help the pharmaceutical industry understand if their products are fit for their intended use. Where the supplier is prepared to share knowledge and formal documents confirming compliance, it is not unreasonable to follow the principles of Quality by Design (QbD) and leverage this knowledge during the risk assessment process.
Furthermore, the drug product manufacturer understands how a material undergoing change interacts with their drug product during manufacture or storage to end of shelf-life. This knowledge, which should be used to assess the probability of the risk event from occurring to an extent that would adversely impact patient safety, might consist of leachable studies conducted previously or theoretical factors (see Tables 2 and 3 in risk assessment). The risk assessment process should follow the same philosophy as what is recommended for new products.
Risk Control
After the risk assessment, as for new products, risk control activities can be performed. Depending on the assessment, these activities can include collection of further information from a supplier, use of prior knowledge, performing extraction or leachables studies, and if needed further safety assessments, to mitigate and accept the change. (see knowledge gathering and safety assessment pages)
Examples
For example, based on risk assessment, a company may decide that a change to the material of construction of the rubber stopper for its product is a major change. The company may then evaluate existing extractables data from the supplier. This may be done according to known protocols/concepts such as those described in USP.[12], [13], [14], [15] Further extractables studies may be needed which can be done by the company or supplier and can include comparisons between the two materials. Extractables profiles of the new and current material can be evaluated with respect to potential patient exposure to leachables.
If the potential patient exposure is not within acceptable levels, process or product specific leachable or simulation studies might be conducted in addition to routine stability studies using pre and post-change rubber stoppers to support and justify the change. The complexity of the study design depends on the variables that might occur and should be evaluated case by case. The study design should be justified and documented.
As another example, a company may decide that a change of the composition of the siliconization solution used for the drug primary container has a medium impact on the potential leachables profile. In this case, the company may opt to conduct a paper-based risk control evaluation and thus documentation and data provided by the vendor are important. In some cases, a comparative leachables study might be planned.
A third example is single use systems where changes have been supported by a paper-based calculation using worst case extractables to complement other risk controls such as clearance or purging, and to decide on the need for additional studies. If the change includes a material that has been used in other products, prior knowledge such as supplier information, and studies done on the material can be used in risk control.
Finally, for minor impacts. risk control may include document review. As an example, a change to a lower gamma irradiation energy can be considered a best-case with respect to the former situation, and therefore no further extractables or leachables study is needed on the new material (or product) to accept the change. This evaluation could then be described in the change control record and relevant quality documentation generated.
Lifecycle Management Documentation
The risk management process should be documented within the applicable quality system as per company procedures. This could include the potential impact of the change to, e.g., a QTPP (for extractables and leachables). The applicable risk assessment methodology/tool (e.g., FMEA, other) should be included and/or referenced.
As illustrated in Figure 1, the outcome of the risk assessment is either risk acceptance, risk reduction (mitigation), or a decision that the risk is unacceptable. Each of these situations can be documented with decisions described. For example, acceptance of risk related to leachables should be documented and the company can proceed with the change as per the product quality system. In the case where risk control activities to reduce risk is required, study or other risk control activity conclusions and decisions should be documented, and the change established as per the applicable PQS activities. If the risk is unacceptable, e.g., change is not supported by the QTPP, or other product standard, the rationale should be documented as per the product quality system.
As noted earlier, depending on the type of change (i.e., relative risk), the documentation will also include submission of specific regulatory updates and supplements.
References
[1] An Industry Proposal for Change Notification Practices for Single-Use Biomanufacturing Systems. BPOG. BPSA. https://www.biophorum.com/wp-content/uploads/2017/05/Change-notification-practices-for-single-use-systems.pdf
[2] ICH Q8 (R2), Pharmaceutical Development. 2009
[3] US FDA. Guidance for Industry, PAT — A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. 2004
[4] US FDA. Pharmaceutical cGMPs for the 21st Century – a Risk-based Approach. Final Report. 2004. https://www.fda.gov/media/77391/download
[5] ICH Q12, Final version adopted 20 November 2019 (Step4), Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management
[6] European Medicines Agency post-authorisation procedural advice for users of the centralised procedure, 20 December 2019, EMEA-H-19984/03 Rev. 84, Human Medicines Evaluation Division
[7] Commission Regulation (EC) No 1234/2008, Annex II, Classification of variations
[8] Guidance for industry. Changes to an approved NDA or ANDA, Center for Drug Evaluation and Research (CDER), April 2004 (revision 1), http://www.fda.gov/cder/guidance/index.htm
[9] Container Closure Systems for Packaging Human Drugs and Biologics. Guidance for Industry. US Food and Drug Administration. 1999. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/container-closure-systems-packaging-human-drugs-and-biologics
[10] Guideline on Plastic Immediate Packaging Materials. European Medicines Agency. 2005. https://www.ema.europa.eu/en/plastic-primary-packaging-materials
[11] For example, see Pang J, Blanc T, et al. Recognition and identification of UV-absorbing leachables in EPREX pre-filled syringes: an unexpected occurrence at a formulation-component interface. PDA J Pharm Sci Technol. Nov-Dec 2007;61(6):423-32
[12] USP <381> Elastomeric Closures for Injections
[13] USP<661.1> Plastic Materials of Construction
[14] USP <661.2> Plastic Packaging Systems for Pharmaceutical Use
[15] USP <1663> Assessment of Extractables Associated with Pharmaceutical Packaging and Delivery Systems